Rev. chil. radiol. vol.26 no.4 Santiago dic. 2020
INTRODUCCIÓN
También conocida como reumatismo del desierto o fiebre del valle, la coccidioidomicosis es una micosis producida por los hongos dimórficos Coccidioides immitis o C. posadassi1. La primera especie se limita al estado de California (EE.UU.) y ciertas regiones del norte de México; mientras que la segunda se distribuye en las restantes zonas del continente americano2. La principal área endémica de la coccidioidomicosis está localizada en las zonas áridas del sudoeste de los Estados Unidos, y su prevalencia máxima se da en el Valle de San Joaquín, California, por lo que también se le conoce como Fiebre de San Joaquín3.
En México hay tres grandes zonas endémicas de Coccidioides immitis: la zona norte que incluye el norte de Baja California, Sonora, Chihuahua, Nuevo León y Tamaulipas; la zona litoral del Pacífico que se extiende hasta Guerrero e incluye partes de Sonora, Sinaloa, Nayarit y Jalisco; y la zona central que se extiende por Coahuila, Nuevo León, Durango y San Luís Potosí1. En Centroamérica se han reportado casos en el Valle de Montagua en Guatemala y en Comayagua, Honduras. En Sudamérica se han registrado casos en el noreste de Venezuela, en las pampas abiertas del gran Chaco en Paraguay, en las zonas áridas de la Patagonia central en Argentina, en los departamentos de Guajira y Magdalena en Colombia y en los estados de Piauí, Bahía, Ceará y Maranhao, al noreste de Brasil3.
CASO CLÍNICO
Hombre de 20 años de edad, residente de Tijuana hace nueve meses. Inicia su padecimiento hace ocho meses, presentando adeanopatía cervical, la cual progresa a un absceso. Requirió ser intervenido de forma externa, con resección de adenopatía y drenaje de absceso, sin envío a patología. Un mes posterior a esto, inicia con dolor en articulación de la rodilla izquierda, con aumento de volumen progresivo, hipertermia, picos febriles, por lo que es valorado por el Servicio de Traumatología con el diagnóstico de probable artritis séptica. Tratado con antibióticos, sin respuesta al tratamiento; evolucionando a una lesión exofítica, fungante, secretante, y que condiciona imposibilidad de deambulación e inestabilidad articular. Acude a un centro de atención hospitalaria, donde se le realizan estudios de laboratorio: BH con hb de 5.40, hto 18.80, PLT 515,000, neutrófilos segmentados 78%, linfocitos 10%, HIV negativo, DHL 471, VDRL negativo, fosfatasa alcalina 193 (Figuras 1, 2, 3 y 4).

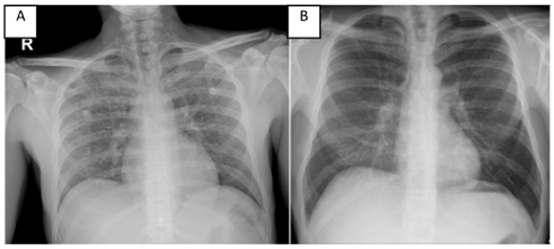
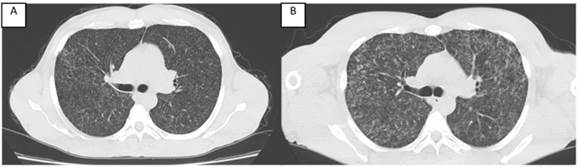
Figura 1: a) Radiografía de tórax AP con patrón reticulonodular bilateral. b) Tomografía de tórax coronal que concuerda con el patrón reticulonodular.

Figura 2: Radiografía AP (a) y lateral (b) de rodilla con colección que condiciona aumento de volumen (cabezas de flecha) y se asocia a cambios de osteólisis de las superficies óseas (Flecha).

Figura 3: Resonancia Magnética en a) secuencia T1, b) secuencia T2 y c) contraste. Se observa colección heterogénea (estrella) en la rodilla con fistulización a la superficie anterior cutánea (cabeza de flecha), osteólisis de las superficies óseas (flecha) e importante realce.

Figura 4: Reconstrucción curva, en coronal en fase contrastada, que evidencia la extensión de la colección (flecha) y la relacionada erosión ósea en la rodilla (estrella).
A la exploración física se encuentra edad cronológica igual a la aparente, con facies dolorosas. Se encuentra al paciente diaforético, cooperador, con Glasgow de 15, palidez de piel y tegumentos y mucosa oral deshidratada. A nivel del cuello, adenomegalias palpables. Se palpan ganglios axilares bilaterales. Presencia de lesión abierta en cara anterior de la rodilla, con salida de coágulos y material seropurulento. Úlcera posterior acompañada de ganglios poplíteos. Fuerza muscular 5/5 de la escala de Daniels, con ROTS normales, llenado capilar de dos segundos, con edema (-) y signo de godete (-).
Se realiza biopsia de ganglio inguinal izquierdo y de lesión de la rodilla derecha, así, como toma de biopsia el 19/06/19. Reporte de proceso infeccioso asociado a coccidioidomicosis.
DISCUSIÓN
Los casos de coccidioidomicosis se asocian a climas áridos y semiáridos, suelos con alto contenido de sales y pH alcalino y vegetación con predominancia de mezquites, cactáceas y gobernadora1. Aunque en su mayoría los casos no se asocian a brotes, estos se producen ocasionalmente cuando se remueven grandes cantidades de suelo, por lo que se han reportado brotes entre militares en entrenamiento, trabajadores arqueológicos, trabajadores granjas de energía solar, y personas expuestas a terremotos y tormentas de polvo4. Los brotes se asocian, además, a los ciclos de abundantes lluvias de febrero a marzo y a las sequías posteriores, teniendo un incremento en la incidencia durante el otoño1.
Estadísticamente, la enfermedad puede afectar a individuos de cualquier edad. En niños muy pequeños y en ancianos frecuentemente tiene una evolución desfavorable. La mayoría de los casos ocurre en varones, probablemente debido a su exposición ocupacional a polvo contaminado; sin embargo, los varones también presentan una mayor tendencia a la diseminación, lo que sugiere un componente genético u hormonal5. La infección se ha informado en todos los grupos etarios; sin embargo, es más frecuente entre campesinos y trabajadores que emigran temporalmente a áreas endémicas; así como en arqueólogos, antropólogos y trabajadores de la construcción6.
Los factores de riesgo son: pacientes VIH-positivos, con recuentos de células T CD4-positivas inferiores a 150/μL; administración de corticosteroides en altas dosis o durante lapsos prolongados; receptores de trasplantes de órganos, enfermos con afecciones malignas, en especial de los órganos linfáticos, en estadios III o IV o sometidos a tratamientos antiblásticos o inmunosupresores; diabetes mellitus; insuficiencia renal crónica en hemodiálisis; embarazo (en especial en los primeros trimestres); el sexo, las formas progresivas son más comunes en los varones después de los 40 años, la raza, los blancos caucásicos son más resistentes a la infección que los afroamericanos, los indígenas y los descendientes de asiáticos. Se ha confirmado la existencia de contagio interhumano al recibir un órgano de un dador infectado2.
La forma infectante de Coccidioides spp. es el artroconidio que forma parte de la fase micelial y que, al desplazarse por el viento, principalmente después de tormentas de polvo, puede ser inhalado por el hospedero7. La coccidioidomicosis no es considerada una enfermedad infecciosa que deba notificarse obligatoriamente y su coexistencia con la tuberculosis en regiones endémicas para ambos padecimientos conlleva un alto grado dificultad diagnóstica7,8.
La infección por coccidioidomicosis presenta los siguientes estadios:
Primoinfección pulmonar asintomática o subclínica2.
Secuelas de la primoinfección pulmonar: manifestaciones semejantes a las de una gripe o una neumonía2.
Neumopatía persistente: perdura más de 6 semanas. Son infiltrados focales o reticulonodulillares2.
Coccidioidomicosis pulmonar crónica excavada: Se presenta en adultos del sexo masculino de más de 40 años y con patologías pulmonares previas, en especial enfermedad pulmonar obstructiva crónica (EPOC)2.
Coccidioidomicosis diseminada subaguda o crónica: La diseminación de la infección ocurre en 1% o menos de los casos8. Se asocia con mayor frecuencia con compromiso cutáneo, ganglionar, óseo y del sistema nervioso central2. La piel, las meninges y el sistema esquelético son los sitios más frecuentemente afectados en los procesos diseminados9. Las lesiones cutáneas son nódulos ulcerados con fondo vegetante, que tienden a crecer en forma excéntrica, úlceras cutáneas de fondo granulomatoso rojizo y bordes socavados o lesiones vegetantes, con un centro parcialmente cicatrizado y borde elevado e invasor. Los ganglios sufren habitualmente un proceso de reblandecimiento supurativo o caseoso que conduce a la fistulización y ulceración de la piel vecina2.
La lesión ósea está presente en 10-50% de los casos diseminados y puede ser monostótica o poliostótica. Las localizaciones óseas más frecuentes son columna vertebral, huesos del cráneo, huesos de manos y pies, tibia y fémur (tercio distal) y pelvis. Las lesiones afectan principalmente la metáfisis y epífisis de huesos largos, pero la diáfisis puede estar comprometida en los huesos cortos de las manos y pies. La lesión articular está constituida por proyecciones papilares con células espumosas y macrófagos con hemosiderina, similar a la observada en la sinovitis villonodular y puede involucrar al cartílago articular y al hueso. Los tejidos blandos periarticulares pueden participar como sitios primarios de infección o estar secundariamente afectados por lesiones articulares y óseas que producen trayectos fistulosos9.
Las lesiones óseas son focos de osteomielitis crónica de localizaciones diversas, predominan en las rodillas, los codos y las vértebras. En los casos más avanzados hay hallazgos radiológicos de osteólisis, periostitis hipertrófica, colecciones purulentas en los tejidos blandos y, por último, fistulización hacia la piel. El compromiso del sistema nervioso se manifiesta con una meningoencefalitis crónica2.
Aunque no existen lesiones radiográficas patognomónicas, se ha descrito que la CM ósea puede presentarse con cinco patrones más frecuentes:
Quistes uniloculados o multiloculados con poca o ninguna reacción ósea.
Erosión marginal con lesiones destructivas en sacabocados, con o son proliferación ósea activa.
Lesiones destructivas extensas con formación de hueso periosteal.
Lesiones de tipo osteomielíticas
Lesiones articulares con participación exclusiva sinovial, o bien con destrucción subarticular y secuestro óseo y formación de abscesos en tejidos blandos9.
El uso de la radiografía como primer estudio de imagen aporta información anatómica y detecta alteraciones que puedan influir para la selección de otros estudios. La ventaja de la Resonancia Magnética es la definición anatómica de los tejidos blandos y óseos que ayuda a definir la extensión de la afectación10.
CONCLUSIÓN
La coccidioidomicosis es una infección sistémica que se presenta en zonas endémicas, por eso, para el tratamiento, es importante relacionar los antecedentes de viajes o estancias de los pacientes en dichas zonas. Esta infección puede evolucionar a coccidioidomicosis diseminada en un muy bajo porcentaje de los casos, en lo que la afectación sistémica puede presentarse a nivel pulmonar, ganglionar, osteomuscular, cutáneo y del sistema nervioso central. En nuestro caso, el paciente tuvo afectación a nivel pulmonar y ganglionar con respuesta paulatina al tratamiento y, a nivel osteomuscular, afectación de la rodilla con compromiso de la función articular y riesgo de mayor compromiso óseo, sin respuesta favorable, por lo que se decidió su desarticulación.
.jpg)